RNAseq with GSNAP
Overview
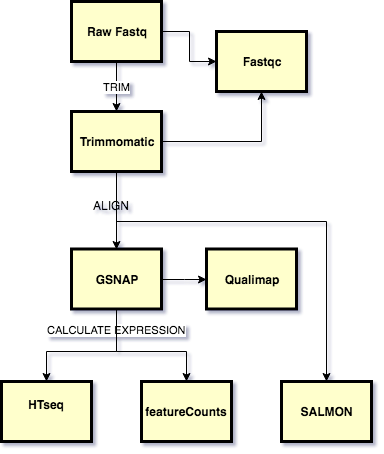
This tutorial shows how to run a standard predefined RNA-seq analysis on the Brown HPC cluster OSCAR, using the bioflows tool. A visual overview of the workflow is shown below
Getting Started
Basic workflow
The workflow consists of the following programs run sequentially on each sample:
- Fastqc: For QC of Raw Fastq reads
- Trimmomatic: Quality and Adapter trimming of raw reads
- Fastqc: Post trimming QC of reads
- GSNAP: alignment of the reads to the reference genome
- Samtools:
- To process alignments:
- convert sam to bam
- remove unmapped reads
- coordinate sort bam
- index bam
- biobambam Suite
- bamsort Sort bams
- bammarkduplicates2 Mark duplicates
- Qualimap QC of the aligments generated
- featureCounts/HTseq Expression quantification based on counting mapped reads
- Salmon: Alignment free quantification of known transcripts
Depending on the user's need the workflow can be adapted for just using a subset of steps or the changes in the sequence as long as they are sensible. We povide a couple of alternative workflows using combinations of the steps in the basic workflow
Steps
The basic steps to running a workflow are:
- Create a control file
- Create your working directory if does not exist, here we assume it is
/users/mydir. - Setup a screen session
The next section provide a short how-to with all the commands to execute the test workflow on Brown University's OSCAR HPCcluster. Once you have the test case working you can implement this on your own data
Prerequisites
Make sure you have access to the OSCAR cluster or request one by contacting support@ccv.brown.edu. If you are not comfortable with the Linux environment, you can consult the tutorial here. You should also set up bioflows.
Danger
You need to have an priority account on OSCAR to run this with real datasets as the resources for exploratory accounts are not sufficient.
Caution
The working directory in a real example can end up being quite large: up to a few terabytes. On OSCAR, you would create the working directory in a location such as your data folder or the scratch folder.
Running the workflow
Create the YAML file
Bioflows uses YAML configuration files to run workflows. A detailed documentation of the YAML file and all the options is shown here. For the current example, copy the following code into a text file and save it in /users/mydir as test_run.yaml.
Note
Edit the parameters in the highlighted lines to change values specific to your username
bioproject: Project_test_localhost
experiment: rnaseq_pilot
sample_manifest:
fastq_file: sample_manifest_min.csv
metadata:
run_parms:
conda_command: source /gpfs/runtime/cbc_conda/bin/activate_cbc_conda
work_dir: /users/mydir
log_dir: logs
paired_end: True
local_targets: False
saga_host: localhost
ssh_user: ccv username
saga_scheduler: slurm
gtf_file: /gpfs/data/cbc/cbcollab/ref_tools/Ensembl_hg_GRCh37_rel87/Homo_sapiens.GRCh37.87.gtf
workflow_sequence:
- fastqc: default
- gsnap:
options:
-d: Ensembl_Homo_sapiens_GRCh37
-s: /gpfs/data/cbc/cbcollab/cbc_ref/gmapdb_2017.01.14/Ensembl_Homo_sapiens_GRCh37/Ensembl_Homo_sapiens_GRCh37.maps/Ensembl_Homo_sapiens.GRCh37.87.splicesites.iit
job_params:
ncpus: 8
mem: 40000
time: 60
- samtools:
subcommand: view
suffix:
input: ".sam"
output: ".bam"
options:
-Sbh:
job_params:
time: 60
- samtools:
subcommand: view
suffix:
input: ".bam"
output: ".mapped.bam"
options:
-bh:
-F: "0x4"
job_params:
time: 60
- samtools:
subcommand: view
suffix:
input: ".bam"
output: ".unmapped.bam"
options:
-f: "0x4"
- bamsort:
suffix:
input: ".mapped.bam"
output: ".srtd.bam"
options:
inputthreads=4:
outputthreads=4:
job_params:
ncpus: 4
mem: 2000
time: 60
- samtools:
subcommand: index
suffix:
input: ".srtd.bam"
job_params:
time: 20
- bammarkduplicates2:
suffix:
input: ".srtd.bam"
output: ".dup.srtd.bam"
job_params:
mem: 2000
time: 60
ncpus: 4
- samtools:
subcommand: index
suffix:
input: ".dup.srtd.bam"
job_params:
time: 20
- qualimap:
subcommand: rnaseq
- htseq-count: default
- featureCounts: default
If you haven't done so already, copy the above into a text file and save it in /users/mydir as test_run.yaml
For this tutorial I have created a small test dataset with 10000 read pairs from human RNAseq data, that's available to all users on OSCAR. It should run within the hour and you should see that all the steps from the workflow have completed.
Create the manifest file
We will now create the sample manifest file, which is in csv format. You can find more information about sample manifest files here. Copy the manifest below into a text file and save it in /users/mydir as sample_manifest_min.csv. As you might notice this is a comma-seperated file.
samp_1299,/gpfs/data/cbc/rnaseq_test_data/PE_hg/Cb2_1.gz,/gpfs/data/cbc/rnaseq_test_data/PE_hg/Cb2_2.gz
samp_1214,/gpfs/data/cbc/rnaseq_test_data/PE_hg/Cb_1.gz,/gpfs/data/cbc/rnaseq_test_data/PE_hg/Cb_2.gz
Run the workflow in a screen session
If you haven't already started a screen session in the setup, start one using the following command:
screen -S rnaseq_tutorial
source /gpfs/runtime/cbc_conda/bin/activate_cbc_conda
bioflows-run test_run.yaml
Workflow outputs
The bioflows-run call will automatically generate several directories, which may or may not have any outputs directed to them depending on which analyses have been run in bioflows. These directories include: sra, fastq, alignments, qc, slurm_scripts, logs, expression, and checkpoints.
For this tutorial you should see the following directories
fastqsymlinks to fastq files.alignmentsSAM and BAM files from GSNAP alignments.qcQC reports from fastqc and qualimap.slurm_scriptsRecords of the commands sent to slurm.logsLog files from various bioflows processes (including the standard error and standard out).expressionExpression values from featureCounts/Salmon/htseq depending on whats specified.checkpointsContains checkpoint records to confirm that bioflows has progressed through each step of the analysis.
Within each folder you will see files that are annotated by samplename_program_suffix. We provide a list of outputs and their contents below
Alternative workflow YAMLs
These YAML's can be used as templates for alternative workflows using various combinations of programs and sequences from the programs defined in the Basic workflow