Period 1 - Standard analyses for RNA seq data (unsupervised)¶
Abstract¶
This workshop summarizes the main approaches to analyzing sequencing data to obtain per-gene count data, but does not go through how to do it explicitly. It demonstrates approaches to reading, annotating, and summarizing count data using tximport and various Bioconductor annotation resources, followed by unsupervised exploratory data analysis. It introduces the (Ranged)SummarizedExperiment data class, variance-stabilizing transformations, and the use of distances to visualize transcriptome shifts through methods such as Principal Components Analysis and Multidimensional Scaling.
This workshop uses quotes and materials from RNA-seq workflow: gene-level exploratory analysis and differential expression by Love, Anders, Kim, and Huber.
Approaches to processing raw data: alignment vs. alignment-free methods¶
The computational analysis of an RNA-seq experiment begins from the FASTQ files that contain the nucleotide sequence of each read and a quality score at each position. These reads are either aligned to a reference genome or transcriptome, or the abundances and estimated counts per transcript can be estimated without alignment. For a paired-end experiment, the software will require both FASTQ files. The output of this alignment step is commonly stored in a file format called SAM/BAM.
RNA-seq aligners differ from DNA aligners in their ability to recognize intron-sized gaps that are spliced from transcripts, and their objective of counting transcripts. Baruzzo et al. 2017 provide a useful summary of software for aligning RNA-seq reads to a reference genome, and benchmark these using synthetic data for base-level and splice junction precision and recall, sensitivity to tuning parameters, and runtime performance. Some popular alignment-based tools include STAR, TopHat2, Subread, and Novoalign. de novo alignment does not use a reference genome: while it offers the possibility of discovering novel transcripts in rearranged genomes and genomes without good references, it requires more resources and manual effort and is less accurate for known transcripts, so for most people working with model organisms it is probably not a good option.
Alignment-free tools have gained popularity for their fast performance and low memory requirements while maintaining good accuracy. Instead of directly aligning RNA-seq reads to a reference genome, they perform "pseudo-alignment" by assigning reads to transcripts that contain compatible k-mers. The most popular of these tools are Salmon and Kallisto. These generate a table of read counts directly without the need for a SAM/BAM file as a starting or intermediate step.
You can easily perform RNA-seq alignment in R using Rsubread and GenomicAlignments, but the rest of this workshop assumes you are starting with the output of some sequence analysis pipeline.
Personally, I would recommend using pseudo-alignment by Salmon according to this tutorial to process raw reads to Transcripts per Million (TPM). It is a very fast, accurate, well-tested, and well-documented command-line tool. It is especially Bioconductor-friendly, because the tximport vignette includes steps to import Salmon results as a DESeqDataSet object for use in the DESeq2 package.
Annotating and importing expression counts¶
Measures of transcript abundance¶
The differential expression analyses introduced here assume you are analyzing a count table of Transcripts Per Million (TPM) or Counts Per Million (CPM) by samples. Note that while TPM and CPM would order the abundance of different transcripts very differently, for differential expression these differences are not as important. You should not use Fragments Per Kilobase per Million reads (FPKM) or otherwise normalize by library size / read depth. The negative binomial log-linear statistical model used by DESeq2 and edgeR are intended for use on count data, and achieve better performance than can be achieved with read depth-normalized data.
Importing with tximport¶
The tximport vignette demonstrates the import of quantification files generated by the Salmon software, but also provides example data and methods of import for other popular RNA-seq quantification software:
library(tximportData)
datadir <- system.file("extdata", package = "tximportData")
list.files(datadir)
Next, we create a named vector pointing to the quantification
files. We will create a vector of filenames first by reading in a
table that contains the sample IDs, and then combining this with dir
and "quant.sf".
samples <- read.table(file.path(datadir, "samples.txt"), header=TRUE)
samples
files <- file.path(datadir, "salmon", samples$run, "quant.sf")
names(files) <- paste0("sample",1:6)
all(file.exists(files))
The tximport package has a single function for importing
transcript-level estimates. The type argument is used to specify
what software was used for estimation ("kallisto", "salmon",
"sailfish", and "rsem" are implemented). A simple list with
matrices, "abundance", "counts", and "length", is returned, where the
transcript level information is summarized to the gene-level. The
"length" matrix can be used to generate an offset matrix for
downstream gene-level differential analysis of count matrices, as
shown below.
Note: While tximport works without any dependencies, it is
significantly faster to read in files using the readr package. If
tximport detects that readr is installed, then it will use the
readr::read_tsv function by default. A change from version 1.2 to
1.4 is that the reader is not specified by the user anymore, but
chosen automatically based on the availability of the readr
package. Advanced users can still customize the import of files using
the importer argument.
The following imports transcript-level counts without summarizing to gene level:
library(tximport)
library(readr)
txi <- tximport(files, type="salmon", txOut=TRUE)
##txi <- tximport(files, type="salmon", tx2gene=tx2gene)
names(txi)
head(txi$counts)
Annotating and summarizing transcripts by gene¶
If your (pseudo-) alignment software provides a map between transcripts and genes, you can specify that map in tximport to summarize counts at the gene level in the same step (see the tx2gene argument of ?tximport::tximport). For this example, such a map is included in the tximportData package and is available via:
tx2gene <- read.csv(file.path(datadir, "tx2gene.csv"))
"In real life" you have a number of options, using either built-in Bioconductor annotations resources or other sources of annotations such as biomaRt or files distributed with read mapping software. Below is the option given in the tximportData package; three more are given in the Appendix.
If you have used an Ensembl transcriptome for (pseudo-)alignment, you can use one of the ensembldb packages to create the transcript-to-gene map.
You can use the following shortcut with ensembldb packages:
suppressPackageStartupMessages(library(EnsDb.Hsapiens.v86))
## the different units you could summarize by:
listColumns(EnsDb.Hsapiens.v86)
tx3gene <- transcripts(EnsDb.Hsapiens.v86, columns="symbol",
return.type="DataFrame")
(tx3gene <- tx3gene[, 2:1])
But there is a more general method that works both for EnsDb databases and for TxDb databases:
library(EnsDb.Hsapiens.v86)
keytypes(EnsDb.Hsapiens.v86)
columns(EnsDb.Hsapiens.v86)
k <- keys(EnsDb.Hsapiens.v86, "TXNAME")
tx4gene <- select(EnsDb.Hsapiens.v86, keys=k,
keytype = "TXNAME", columns = "SYMBOL")
tx4gene <- tx4gene[, 3:2]
# Check that this method is equivalent to the previous one:
all.equal(tx4gene[, 1], tx3gene[, 1])
all.equal(tx4gene[, 2], tx3gene[, 2])
So wherever you get your gene models from, you can convert it to a TxDb and use the same select() commands.
Finally, however you created your transcript to gene map, you can use it to summarize transcripts to genes:
txi.sum <- summarizeToGene(txi, tx2gene)
The SummarizedExperiment data class for representing RNA-seq data¶
The component parts of a SummarizedExperiment object.
assay(se)orassays(se)$countscontains the matrix of countscolData(se)may contain data about the columns, e.g. patients or biological unitsrowData(se)may contain data about the rows, e.g. genes or transcriptrowRanges(se)may contain genomic ranges for the genes/transcriptsmetadata(se)may contain information about the experiment
<img src="https://github.com/waldronlab/BrownCOBRE2018/blob/master/notebooks_day2/SummarizedExperiment.jpg?raw=true", alt="SummarizedExperiment image", style="width:500px">
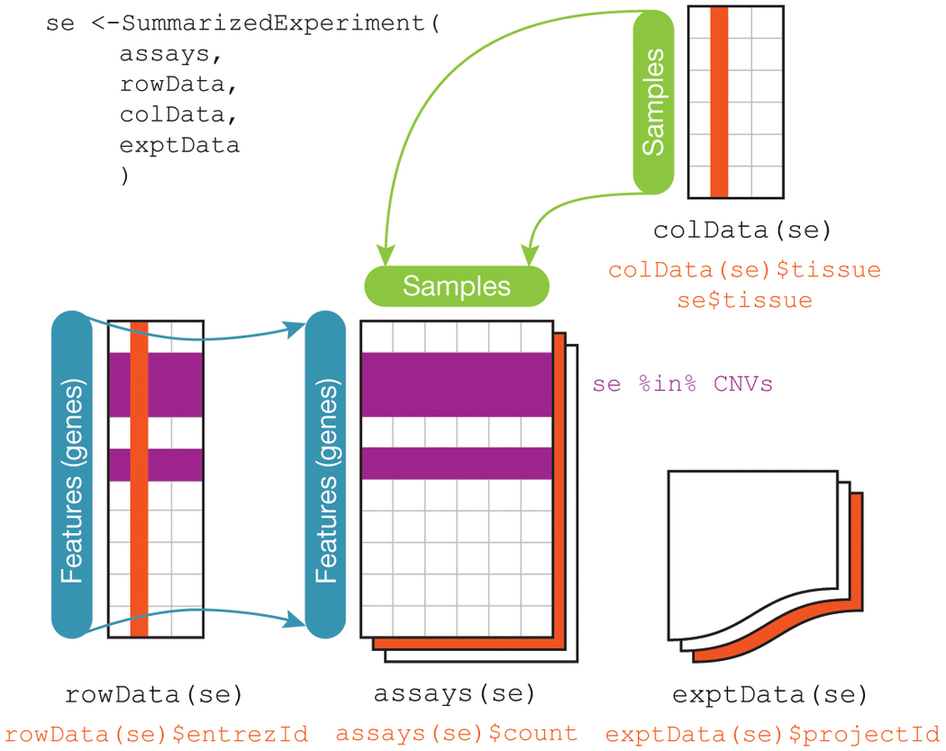{kind=link}
Note: The rowRanges could be a GRanges providing ranges of genes, or a GRangesList providing ranges of exons grouped by gene. Both are accessible from TxDb databases.
RangedSummarizedExperiment is a subclass of SummarizedExperiment, where the rowRanges is not empty. You can convert a SummarizedExperiment to a RangedSummarizedExperiment just by setting its rowRanges.
Some exploratory analysis of a demo RangedSummarizedExperiment:
suppressPackageStartupMessages({
library(airway)
library(SummarizedExperiment)
})
data(airway)
airway
dim(airway)
assayNames(airway)
head(assay(airway), 3)
colSums(assay(airway))
The rowRanges here are a GRangesList. The first gene has 17 exons:
rowRanges(airway)
The rowRanges also contains information about the gene model in the metadata slot. Here we use a helpful R function, str, to display the metadata compactly:
str(metadata(rowRanges(airway)))
The colData:
colData(airway)
Exercise¶
Create your own SummarizedExperiment and RangedSummarizedExperiment objects using the examples in ?SummarizedExperiment. SummarizedExperiment objects can be the starting point for many analyses.
Explore the components of the created objects, including how many rows/columns they have, and what the rowRanges contain.
Exploratory data analysis¶
Standardization / transformation of count data for visual exploration¶
Features (e.g. genes) are typically "standardized", because:
- the differences in overall levels between features are often not due to biological effects but technical ones, e.g. GC bias, PCR amplification efficiency, ...
- otherwise a few very high-variance genes can dominate the distance, and make the distances less biologically relevant
Standardization traditionally meant converting to z-score:
$$x_{gi} \leftarrow \frac{(x_{gi} - \bar{x}_g)}{s_g}$$
But DESeq2 and edgeR provide their own RNA-seq specific standardization methods.
We will use standardization for the purposes of visualization below, but for statistical testing in the next period, we will go back to the original count data as required by the statistical procedures used.
Exploring the airway dataset¶
Once we have our fully annotated SummarizedExperiment object, we can construct a DESeqDataSet object from it that will then form the starting point of the analysis. We add an appropriate design for the analysis (more on model formulae in the next period):
suppressPackageStartupMessages(library(DESeq2))
dds <- DESeqDataSet(airway, design = ~ cell + dex)
Filtering genes not satisfying a minimum expression threshold¶
Our count matrix contains many rows with only zeros, and additionally many rows with only a few fragments total. In order to reduce the size of the object, and to increase the speed of our functions, we can remove the rows that have no or nearly no information about the amount of gene expression. Here we apply the most minimal filtering rule: removing rows of the DESeqDataSet that have no counts, or only a single count across all samples. Additional weighting/filtering to improve power is applied at a later step in the workflow.
nrow(dds)
dds <- dds[ rowSums(counts(dds)) > 1, ]
nrow(dds)
Note: For differential expression analysis later, filtering is
allowable but not necessary if using Independent Hypothesis Weighting,
as is the default behavior of DESeq2. Independent hypothesis weighting (IHW) is a multiple testing procedure that increases power compared to the method of Benjamini and Hochberg by assigning data-driven weights to each hypothesis.
The rlog and variance stabilizing transformations¶
Many common statistical methods for exploratory analysis of multidimensional data, for example clustering and principal components analysis (PCA), work best for data that generally has the same range of variance at different ranges of the mean values. When the expected amount of variance is approximately the same across different mean values, the data is said to be homoskedastic. For RNA-seq counts, however, the expected variance grows with the mean. For example, if one performs PCA directly on a matrix of counts or normalized counts (e.g. correcting for differences in sequencing depth), the resulting plot typically depends mostly on the genes with highest counts because they show the largest absolute differences between samples. A simple and often used strategy to avoid this is to take the logarithm of the normalized count values plus a pseudocount of 1; however, depending on the choice of pseudocount, the genes with the very lowest counts will contribute a great deal of noise to the resulting plot, because taking the logarithm of small counts actually inflates their variance. We can quickly show this property of counts with simulated data (here, Poisson counts with a range of lambda from 0.1 to 100). We plot the standard deviation of each row (genes) against the mean:
options(repr.plot.height=3, repr.plot.width=5)
lambda <- 10^seq(from = -1, to = 2, length = 1000)
cts <- matrix(rpois(1000*100, lambda), ncol = 100)
library(vsn)
meanSdPlot(cts, ranks = FALSE)

And for logarithm-transformed counts:
options(repr.plot.height=3, repr.plot.width=5)
log.cts.one <- log2(cts + 1)
meanSdPlot(log.cts.one, ranks = FALSE)

The logarithm with a small pseudocount amplifies differences when the values are close to 0. The low count genes with low signal-to-noise ratio will overly contribute to sample-sample distances and PCA plots.
As a solution, DESeq2 offers two transformations for count data that stabilize the variance across the mean: the regularized-logarithm transformation or rlog (Love, Huber, Anders, Genome Biology 2014), and the variance stabilizing transformation (VST) for negative binomial data with a dispersion-mean trend (Anders and Huber, Genome Biology 2010), implemented in the vst function.
For genes with high counts, the rlog and VST will give similar result to the ordinary log2 transformation of normalized counts. For genes with lower counts, however, the values are shrunken towards the genes' averages across all samples. The rlog-transformed or VST data then becomes approximately homoskedastic, and can be used directly for computing distances between samples, making PCA plots, or as input to downstream methods which perform best with homoskedastic data.
Which transformation to choose? The rlog tends to work well on
small datasets (n < 30), sometimes outperforming the VST when there is
a large range of sequencing depth across samples (an order of
magnitude difference). The VST is much faster to compute and is less
sensitive to high count outliers than the rlog. We therefore recommend
the VST for large datasets (hundreds of samples). You can perform both
transformations and compare the meanSdPlot or PCA plots generated,
as described below.
Note that the two transformations offered by DESeq2 are provided for applications other than differential testing. For differential testing we recommend the DESeq function applied to raw counts, as described later in this workflow, which also takes into account the dependence of the variance of counts on the mean value during the dispersion estimation step.
The function rlog returns an object based on the SummarizedExperiment class that contains the rlog-transformed values in its assay slot.
options(repr.plot.height=3, repr.plot.width=5)
rld <- rlog(dds, blind = FALSE)
head(assay(rld), 3)
meanSdPlot(assay(rld), ranks=FALSE)

The function vst returns a similar object:
vsd <- vst(dds, blind = FALSE)
head(assay(vsd), 3)
meanSdPlot(assay(vsd), ranks=FALSE)

In the above function calls, we specified blind = FALSE, which means
that differences between cell lines and treatment (the variables in
the design) will not contribute to the expected variance-mean trend of
the experiment. The experimental design is not used directly in the
transformation, only in estimating the global amount of variability in
the counts. For a fully unsupervised transformation, one can set
blind = TRUE (which is the default).
To show the effect of the transformation, in the figure below
we plot the first sample
against the second, first simply using the log2 function (after adding
1, to avoid taking the log of zero), and then using the rlog- and VST-transformed
values. For the log2 approach, we need to first estimate size factors to
account for sequencing depth, and then specify normalized=TRUE.
Sequencing depth correction is done automatically for the rlog
and the vst.
dds <- estimateSizeFactors(dds)
ddsESF <- estimateSizeFactors(dds)
df1 <- data.frame(log2(counts(ddsESF, normalized=TRUE)[, 1:2] + 1))
df1$transformation <- "log2(x + 1)"
df2 <- data.frame(assay(rld)[, 1:2])
df2$transformation <- "rld"
df3 <- data.frame(assay(vsd)[, 1:2])
df3$transformation <- "vsd"
df <- rbind(df1, df2, df3)
colnames(df)[1:2] <- c("x", "y")
head(df)
options(repr.plot.width=6, repr.plot.height=2.5)
library(ggplot2)
ggplot(df, aes(x = x, y = y)) + geom_hex(bins = 80) +
coord_fixed() + facet_grid( . ~ transformation)

Scatterplot of transformed counts from two samples. Shown are scatterplots using the log2 transform of normalized counts (left), using the rlog (middle), and using the VST (right). While the rlog is on roughly the same scale as the log2 counts, the VST has a upward shift for the smaller values. It is the differences between samples (deviation from y=x in these scatterplots) which will contribute to the distance calculations and the PCA plot.
We can see how genes with low counts (bottom left-hand corner) seem to be excessively variable on the ordinary logarithmic scale, while the rlog transform and VST compress differences for the low count genes for which the data provide little information about differential expression.
Boxplots of transformed distributions¶
Boxplots of the count distributions in each sample are a good way to understand the effects these transformations have.
par(mfrow=c(1,3))
boxplot(log2(assay(ddsESF)+1), las=2, main="log2(x+1)")
boxplot(assay(rld), las=2, main="rld")
boxplot(assay(vsd), las=2, main="vsd")

Sample distances¶
The importance of distances¶
High-dimensional data are complex and impossible to visualize in raw form. They represent thousands of dimensions, but we can only visualize 2-3.

All clustering and classification of samples and/or genes involves combining or identifying objects that are close or similar. Distances or similarities are mathematical representations of what we mean by close or similar. The choice of distance is important, varies for different subject areas and types of data.
See: http://master.bioconductor.org/help/course-materials/2002/Summer02Course/Distance/distance.pdf
Distances for exploratory RNA-seq data analysis¶
A useful first step in an RNA-seq analysis is often to assess overall similarity between samples: Which samples are similar to each other, which are different? Does this fit to the expectation from the experiment's design?
We use the R function dist to calculate the Euclidean distance between samples. To ensure we have a roughly equal contribution from all genes, we use it on the rlog-transformed data. We need to transpose the matrix of values using t, because the dist function expects the different samples to be rows of its argument, and different dimensions (here, genes) to be columns.
sampleDists <- dist(t(assay(rld)))
sampleDists
We visualize the distances in a heatmap in a figure below, using the function pheatmap from the pheatmap package.
library(pheatmap)
library(RColorBrewer)
In order to plot the sample distance matrix with the rows/columns
arranged by the distances in our distance matrix,
we manually provide sampleDists to the clustering_distance
argument of the pheatmap function.
Otherwise the pheatmap function would assume that the matrix contains
the data values themselves, and would calculate distances between the
rows/columns of the distance matrix, which is not desired.
We also manually specify a blue color palette using the
colorRampPalette function from the RColorBrewer package.
options(repr.plot.height=3, repr.plot.width=5)
sampleDistMatrix <- as.matrix( sampleDists )
rownames(sampleDistMatrix) <- paste( rld$dex, rld$cell, sep = " - " )
colnames(sampleDistMatrix) <- NULL
colors <- colorRampPalette( rev(brewer.pal(9, "Blues")) )(255)
pheatmap(sampleDistMatrix,
clustering_distance_rows = sampleDists,
clustering_distance_cols = sampleDists,
col = colors)

Heatmap of sample-to-sample distances using the rlog-transformed values.
Note that we have changed the row names of the distance matrix to contain treatment type and patient number instead of sample ID, so that we have all this information in view when looking at the heatmap.
Another option for calculating sample distances is to use the
Poisson Distance, implemented in the
PoiClaClu CRAN package.
This measure of dissimilarity between counts
also takes the inherent variance
structure of counts into consideration when calculating the distances
between samples. The PoissonDistance function takes the original
count matrix (not normalized) with samples as rows instead of columns,
so we need to transpose the counts in dds.
library(PoiClaClu)
poisd <- PoissonDistance(t(counts(ddsESF)))
We plot the heatmap below.
options(repr.plot.height=3, repr.plot.width=5)
samplePoisDistMatrix <- as.matrix( poisd$dd )
rownames(samplePoisDistMatrix) <- paste( rld$dex, rld$cell, sep=" - " )
colnames(samplePoisDistMatrix) <- NULL
pheatmap(samplePoisDistMatrix,
clustering_distance_rows = poisd$dd,
clustering_distance_cols = poisd$dd,
col = colors)

Heatmap of sample-to-sample distances using the Poisson Distance.
PCA plot¶
Another way to visualize sample-to-sample distances is a principal components analysis (PCA). In this ordination method, the data points (here, the samples) are projected onto the 2D plane such that they spread out in the two directions that explain most of the differences (figure below). The x-axis is the direction that separates the data points the most. The values of the samples in this direction are written PC1. The y-axis is a direction (it must be orthogonal to the first direction) that separates the data the second most. The values of the samples in this direction are written PC2. The percent of the total variance that is contained in the direction is printed in the axis label. Note that these percentages do not add to 100%, because there are more dimensions that contain the remaining variance (although each of these remaining dimensions will explain less than the two that we see).
plotPCA(rld, intgroup = c("dex", "cell"))

PCA plot using the rlog-transformed values. Each unique combination of treatment and cell line is given its own color.
Here, we have used the function plotPCA that comes with DESeq2.
The two terms specified by intgroup are the interesting groups for
labeling the samples; they tell the function to use them to choose
colors. We can also build the PCA plot from scratch using the
ggplot2.
This is done by asking the plotPCA function
to return the data used for plotting rather than building the plot.
See the ggplot2 documentation
for more details on using ggplot.
pcaData <- plotPCA(rld, intgroup = c( "dex", "cell"), returnData = TRUE)
pcaData
percentVar <- round(100 * attr(pcaData, "percentVar"))
We can then use these data to build up a second plot in a figure below, specifying that the color of the points should reflect dexamethasone treatment and the shape should reflect the cell line.
ggplot(pcaData, aes(x = PC1, y = PC2, color = dex, shape = cell)) +
geom_point(size =3) +
xlab(paste0("PC1: ", percentVar[1], "% variance")) +
ylab(paste0("PC2: ", percentVar[2], "% variance")) +
coord_fixed()

PCA plot using the rlog-transformed values with custom ggplot2 code. Here we specify cell line (plotting symbol) and dexamethasone treatment (color).
From the PCA plot, we see that the differences between cells (the
different plotting shapes) are considerable, though not stronger than the differences due to
treatment with dexamethasone (red vs blue color). This shows why it will be important to
account for this in differential testing by using a paired design
("paired", because each dex treated sample is paired with one
untreated sample from the same cell line). We are already set up for
this design by assigning the formula ~ cell + dex earlier.
MDS plot¶
Another plot, very similar to the PCA plot, can be made using the multidimensional scaling (MDS) function in base R. This is useful when we don't have a matrix of data, but only a matrix of distances. Here we compute the MDS for the distances calculated from the rlog transformed counts and plot these in a figure below.
MDS plot using rlog-transformed values.
mds <- cbind(as.data.frame(colData(rld)), cmdscale(sampleDistMatrix))
ggplot(mds, aes(x = `1`, y = `2`, color = dex, shape = cell)) +
geom_point(size = 3) + coord_fixed()

In the figure below we show the same plot for the PoissonDistance.
MDS plot using the Poisson Distance.
mdsPois <- cbind(as.data.frame(colData(ddsESF)),
cmdscale(samplePoisDistMatrix))
ggplot(mdsPois, aes(x = `1`, y = `2`, color = dex, shape = cell)) +
geom_point(size = 3) + coord_fixed()

Exercise: The following code will read example .bam files containing only reads from human chromosome 14, and count overlaps with known genes on chromosome 14. Follow these steps, then perform exploratory analysis on the resulting data object
library(RNAseqData.HNRNPC.bam.chr14) bfp = RNAseqData.HNRNPC.bam.chr14_BAMFILES library(Rsamtools) bfl = BamFileList(file=bfp) library(Homo.sapiens) gr14 = genes(Homo.sapiens, filter=list(tx_chrom="chr14"), columns="SYMBOL") library(GenomicAlignments) rse <- summarizeOverlaps(gr14, bfl)
Appendix ¶
Additional options for mapping transcripts to genes¶
Option 2. Bioconductor TxDb.* packages, which are available for a number of model species (search for "TxDb" at https://bioconductor.org/packages/3.6/data/annotation/). These are slightly less convenient for this purpose than the Ensembl packages. For example, there is a TxDb package Mus Musculus from UCSC build mm10 based on the knownGene Track:
library(TxDb.Mmusculus.UCSC.mm10.knownGene)
txdb <- TxDb.Mmusculus.UCSC.mm10.knownGene
keytypes(txdb)
k <- keys(txdb, "TXNAME")
columns(txdb)
tx4gene <- select(txdb, keys=k, keytype="TXNAME", column="GENEID")
genes(txdb)
exonsBy(txdb)
Option 3. Find a TxDb from AnnotationHub
This opens up more annotation files from different sources. First find and choose a TxDb database:
suppressPackageStartupMessages(library(AnnotationHub))
ah <- AnnotationHub()
query(ah, "TxDb")
txdb <- ah[["AH52245"]] #TxDb.Athaliana.BioMart.plantsmart22.sqlite
Then use it to create the transcript-gene map:
keytypes(txdb)
columns(txdb)
k <- keys(txdb, "TXNAME")
tx5gene <- select(txdb, keys=k, keytype="TXNAME", column="GENEID")
head(tx5gene)
Option 4. Import a GFF annotation file from somewhere else.
To convert the GFF file these to a TxDb database, use the makeTxDbFromGFF() function from the GenomicFeatures package:
library(GenomicFeatures)
gffFile <- system.file("extdata", "GFF3_files", "a.gff3",
package="GenomicFeatures")
txdb <- makeTxDbFromGFF(file=gffFile,
dataSource="partial gtf file for Tomatoes for testing",
organism="Solanum lycopersicum")
keytypes(txdb)
columns(txdb)
k <- keys(txdb, "TXNAME")
tx6gene <- select(txdb, keys=k, keytype="TXNAME", column="GENEID")
head(tx5gene)
Note, the GenomicFeatures package enables you to make a TxDb from a variety of sources:
grep("makeTxDb", ls("package:GenomicFeatures"), val=TRUE)